An Overview of Galactosemia and Its comparison to Electron Transport Chain and Medium Chain Acyl-CoA Dehydrogenase Deficiency
Any deficiencies of the enzymes in a metabolic pathway will likely lead to a metabolic disorder. Galactosemia is an autosomal recessive disorder of galactose metabolism.1 The disease is caused by the deficiency in galactokiase (GALK), galactose-1-phosphate uridyltransferase (GALT), or UDP-galactose 4’ epimerase.1 The deficiency in GALT is the most common cause among the three.1 The presence of the galactosemia can be detected by the use of newborn screening (NBS), where a positive result indicates the presence of the disease.1
The GALT enzyme catalyzes the double displacement reaction where galactose-1-phosphate and uridine diphosphate glucose (UDP glucose) is converted into uridine diphosphate galactose (UDP galactose) and glucose-1-phosphate in the Leloir pathway (Figure 1).2 The mechanism of this double displacement reaction is also known (Figure 2).3 The GALT enzyme has an imidazole ring which contains a nitrogen atom to act as a nucleophile and attacks UDP-glucose to break the phosphoanhydride bond.3 The phosphate group is transferred from UDP-glucose to the enzyme; therefore enzyme-UMP and glucose-1-phosphate are formed.3 The galactose-1-phosphate uses its oxygen atom in its phosphate group to nucleophilic attack the enzyme-UMP.3 The phosphoanhydride bond in the enzyme is broken and the phosphate group is transferred to galactose-1-phosphate.3 The UDP-galactose and a free GALT are resulted.3 The free enzyme acts as a catalyst and can be used again in the next cycle of reaction.3 The glucose-1-phosphate will then enter glycolysis pathway to be further broken down into two pyruvate molecules with the production of ATP and NADH, the energy reservoirs used in body.2 The UDP-galactose can also synthesize glycoproteins and glycolipids through galactosylation.2 The EC number of GALT enzyme is found out to be 2.7.7.12.4 The structure of GALT is available from Escherichia Coli.4 The GALT enzyme is a dimer that is composed of two identical subunits.4 There are 348 amino acid residues in each subunit with two associated metal ions, iron and zinc.4 Each subunit is made of alternating alpha-beta-alpha secondary structures and forms a half barrel domain (Figure 3).4 The structure is conserved through evolution, thus the GALT enzyme in human is expected to be similar to the GALT in E.coli.5
Galactosemia can be presented in three common forms.1 Classic galactosemia, whose genotype is mutated by changing glutamine to arginine at position 188 (Q188R/Q188R) in exon 6, is the most severe form of the disease.5 In this type of galactosemia, the GALT is completely non-functional and it is lethal if lactose is consumed.1 Due to the absence of GALT activity, galactose will not be successfully converted into glucose.1 Therefore, the level of galactose or galactose-1-phosphate will increase in the patients, who will likely to develop lethal E. coli sepsis, liver problems, and cataracts.1 Long-term diet-independent complications will also likely to be developed, such as cognitive impairments, premature ovarian insufficiency (POL) in female, speech defects and language delay.1 The patients with this kind of disease also show abnormalities in their cerebella peduncles, dentate nuclei and periventricular white matter .1
The other two forms of the disease are clinical and biochemical galactosemia.1 Clinical galactosemia is caused by a change of serine to leucine at position 135 (S135L/S135L). This form is less severe than the classic form but still requires treatments.1 The patients demonstrate a normal breath test on galactose.1 They will also manifest growth failure, liver disease and cataracts, but they do not show POL or long-term diet-independent complications as classic galactosemia.1 Biochemical galactosemia (N314D/Q188R), the third form of this disease, which changes asparagine to aspartic acid at position 314 or glutamine to arginine at position 188, is the least severe variant and often considered to be benign.1 The GALT activity is reduced in these patients but is still detectable.1 The patients still have increased level of galactose or galactose-1-phosphate in the body, however, they normally do not show other symptoms.1
There are some therapies available to relieve the acute symptoms caused by galactosemia. First of all, the patient should be put on lactose-free diet immediately after the disease is detected.1 The affected infants are given soy-based formula instead of lactose-based formula or breast feeding.1 The patients should still be monitored in a long-term for E.coli sepsis and coagulopathy since the long-term diet-independent complications, such as language delay, cognitive impairment and cataracts, are still possible to occur.1
Galactosemia is not a lethal mutation when it is compared with electron transport chain (ETC) mutations despite its acute and chronic clinical manifestations. Both of GALT and ETC are used in carbohydrate metabolism which provides energy to the body, but ETC mutations are often fatal.6 ETC is an important step in the process of pyruvate oxidation under aerobic conditions.6 It takes NADH and FADH2, which are made through glycolysis and tricarboxylic acid cycle (TCA cycle), donates their electrons to oxygen and is followed by production of ATP through ATP synthase.6 The GALT enzyme is used in Leloir pathway to convert galactose into glucose which will then enter the glycolysis pathway, whose products will also undergo ETC under aerobic condition.2 Therefore, ETC is always necessary in the aerobic carbohydrate metabolism whereas the GALT enzyme just functions to bring the galactose into glycolysis in the form of glucose. Galactose is normally obtained from lactose, which is a disaccharide made of a pair of glucose and galactose molecules, but it is not a necessary energy resource as glucose is normally widely available. The metabolism pathway of glucose is independent on GALT activity but it requires ETC to maximize its production of energy.6 And also, NAD+ and FAD are the side products from ETC which are required in glycolysis.6 Therefore, the function of ETC is more important than the GALT enzyme and the mutations in ETC is more severe.
Medium chain acyl-CoA dehydrogenase (MCAD) deficiency is also an autosomal recessive disease as galactosemia.7 The affected individuals have a deficiency of beta-oxidation of fatty acids in the mitochondria.7 The MCAD deficiency can be treated by frequent and consistent ingestion of complex carbohydrates, less fat ingestion, and intake of oral carnitine.7 The treatment of MCAD deficiency is different to the treatment of galactosemia. The patients with galactosemia are put on free-lactose diet to completely avoid the intake of galactose, whereas the patients of MCAD deficiency can still ingest some amount of fat.1,7 As galactosemia, the MCAD deficiency is also not lethal.7 MCAD is used in the pathway in which fatty acids are converted into acetyl-CoA which will then enter TCA cycle.7 The glucose metabolism pathway is not affected by the deficiency in this enzyme therefore the body can still rely on the glucose for energy sources. Like galactose, metabolism of fatty acids is not necessary for survival. The patients can simply reduce the ingestion of galactose and fatty acids to relieve the symptoms of galactosemia and MCAD deficiency, respectively.1, 7
A variety of diseases in different metabolic pathways have already been revealed and their treatments have been widely studied. Some of the disorders are lethal whereas the others are not. Further investigations and analysis of each disease are still needed, as there may be some other variants of the disease that we have not discovered.
References:
Appendix:

Figure 1: Leloir pathway. Galactose is converted into galactose-1-P by galactokinase (GALK) with the consumption of 1 equivalent of ATP. Galactose-1-phosphate uridyltransferase (GALT) converts galactose-1-phosphate to UDP-galactose and UDP-glucose to glucose-1-phosphate. The UDP-galactose is converted back to UDP-glucose by UDP-galactose 4’ epimerase (GALE). UDP-galactose can also enter glycosylation pathways.2
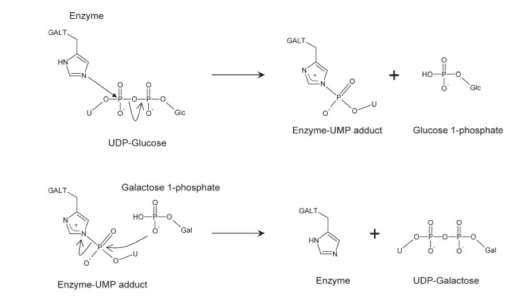
Figure 2: Mechanism of Galactose-1-phosphate uridyltransferase (GALT) in Leloir pathway. The nitrogen in the imidazole ring in GALT enzyme acts as a nucleophile which attacks the phosphate group in UDP-glucose and breaks the phosphoanhydride bond. The UDP-glucose therefore losses a phosphate group to the enzyme and becomes glucose-1-phosphate. The GALT enzyme with the phosphate group (enzyme-UMP) is then nucleophilic attacked by the oxygen in the phosphate group of galactose-1-phosphate. The phosphoanhydride bond in enzyme-UMP is broken therefore the enzyme losses a phosphate group to galactose-1-phosphate which becomes the UDP-galactose. The enzyme can be recycled and used for the next round of reaction.3
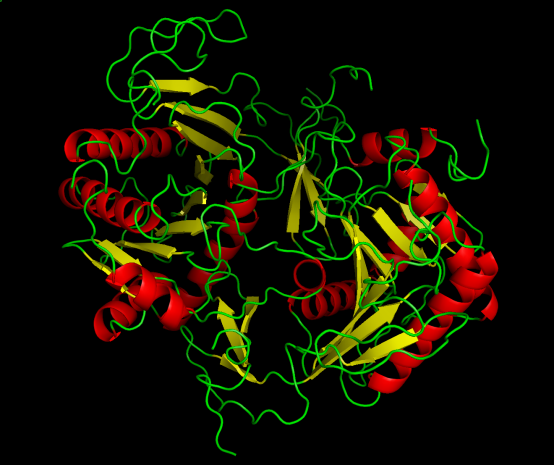
Figure 3: The structure of Galactose-1-phosphate uridyltransferase (GALT) in Escherichia Coli. The alpha helices are represented in red and beta sheets are shown in yellow. The green color shows the amino acid strands connecting the secondary structures. The enzyme is a dimer of two identical subunits. Each subunit exhibits a barrel-like shape and made of 348 amino acids (Retrieved from PDB file: 1HXQ).4
Delivering a high-quality product at a reasonable price is not enough anymore.
That’s why we have developed 5 beneficial guarantees that will make your experience with our service enjoyable, easy, and safe.
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more