First described by David Bloom in 1954, Bloom syndrome (BS) is an autosomal recessive genetic condition resulting in widespread genomic instability and a subsequent predisposition to the development of cancer (Cunniff, 2017). BS is an example of a ‘chromosome breakage syndrome,’ characterised by defective DNA metabolism and genetic repair mechanisms (Duker, 2002).
Effecting less than 300 people worldwide, symptoms include abnormally small size and noticeable redness of the cheeks and nose. People with BS also experience impaired immune system function, reduced fertility and decreased intellectual ability in some cases (Cunniff, 2017). Over a third of the patients in the Bloom syndrome registry descend from Ashkenazi Jewish ethnic lineages (Sanz, 2006).
The underlying genetic landscape of BS is complex and not yet fully understood. This essay will discuss clinical symptoms, molecular aetiology, functional consequences, methods of diagnosis and current treatment options for BS.
As BS is inherited in an autosomal recessive fashion, each parent of an affected individual must carry one copy of the recessive gene even if they themselves do not have BS. In cases where both parents are carriers, there is a 25% chance that their offspring will have the condition, a 50% chance that they will be carriers and a 25% chance that they will not have the condition nor be carriers (Sanz, 2006). BS is known to affect both sexes equally and is common among children resulting from parental consanguinity (German, 2007). As there are so few cases, it is difficult to calculate a specific overall incidence rate (Tikoo, 2010).
The gene underlying the pathology of BS, BLM, was mapped to the locus 15q26.1 by German et al (1994) and is currently the only gene known to cause BS (Cunniff, 2017). It encodes for the RECQL3 protein (also known as BLM), a DNA helicase important in a number of processes relating to DNA repair and replication. There are other conditions caused by defects in the RECQ family of proteins including Werner Syndrome (WS) and Rothmund-Thomson syndrome (RTS) (Duker, 2002). The specific genes involved play unique and distinct roles in the maintenance of genomic integrity, making these conditions distinct from BS. For instance, premature aging is commonly observed in WS and a hallmark facial rash is observed in RTS (Larizza, 2010).
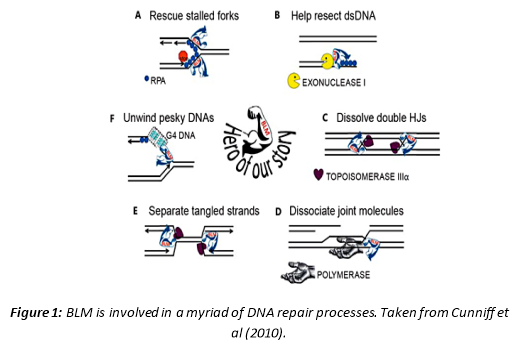
BLM has a huge role in maintaining genomic integrity. Reviewed by Cunniff et al (2017), it participates in several pathways related to the DNA damage response (Figure 1). Described in figure 1 as the ‘hero of the story,’ BLM is instrumental in processing DNA replication intermediates. Some of its duties include recruiting RAD51 and RPA to sites of stalled replication to prevent premature recombination, helping to dissolve double Holliday junctions to limit DNA crossover formation and separating tangled strands that might otherwise lead to errors in recombination. Consequently, BLM mutation can be catastrophic to the cell.
There have been over 60 mutations of the BLM gene identified that are known to cause BS. The most common, BLMAsh, has a carrier frequency of 1 in 110 in the Ashkenazi Jewish population and is unique to patients with this ethnic background (Cunniff, 2017). Ellis et al (1995) performed a mutation analysis of 13 unrelated patients with BS, identifying ten mutations, seven of which were unique. They suggested that the absence of wild-type BLM in turn affects the activity of other DNA repair enzymes, creating a cascade effect.
A 2007 report identified 49 BLM mutations that had not been previously described and represents the most comprehensive study of this kind to date in BS (German, 2007). Nineteen of these mutations were found to recur throughout the population of people with BS. There were also several BLM mutations identified that were unique to just one individual, suggesting that there may still be more as yet unidentified BS-causing BLM mutations. Additionally, in nine of the people studied, no BLM mutation could be detected, highlighting the possibility of BS-causing mutations in genes other than BLM. This study also found that people from similar ethnic backgrounds harbouring the recurrent BLM mutations likely inherited them from a common ancestor or ‘founder.’
The identified mutations included substitutions, deletions and insertions. The most prominent functional consequence was premature translation termination, resulting in incomplete protein product. Amino acid substitution leading to missense mutation was cited as perhaps the most important identified, given that these generally occurred within the DNA helicase region of the gene that is essential for enzymatic activity.
Studies have suggested that heterozygotes with only one pathogenic BLM variant may also be at increased risk of developing cancer while exhibiting no other BS symptoms (Cunniff, 2017).
Cells with mutated BLM are unable to perform double-strand break (DSB) repair and this is a key driver behind the hyper-mutational capability of BS cells (Langland, 2002). Chromosomal mutations also occur at high frequency in BS cells and there is vastly increased chromatid exchange leading to profound genetic instability. These cumulative abnormalities mean BS cells are among the most hyper-mutated known (German, 2007). As a result, people with BS experience a markedly higher incidence of cancer, of any type and often at a young age (Cunniff, 2017). Tikoo et al (2010) suggest that BLM mutation may be central to the ‘common master regulatory step’ that precedes neoplastic transformation in all cancers.
A process known as sister-chromatid exchange (SCE) also occurs at a higher than normal rate in BS cells. During DNA replication, each chromosome becomes two identical ‘sister’ chromatids that are joined with one another. SCE is when sister chromatids break and recombine with each other, allowing exchange of genetic material between the two. A hallmark characteristic of BS cells is a 10x higher frequency of SCE events than normal cells; this allows BS to be distinguished from other similar conditions during diagnosis (Amor-Gueret, 2006; Sanz, 2006). A higher frequency of SCE is indicative of higher rates of mitosis and is a result of abnormal DNA repair attribute to mutated BLM.
In the normal cell, BLM is recruited to DSBs on replication forks – these are structures facilitating the synthesis of new DNA prior to cell division. A primary function of BLM is to remove the blockage caused by a DSB to prevent premature recombination (Amor-Gueret, 2006). This is crucial in helping to suppress tumorigenesis and BLM has been shown to be indispensable for this process, even though there are alternative mechanisms available to the cell involving different proteins e.g. GEN1 and MUS81 (Arora, 2014).
Such widespread genetic instability would normally trigger affected cells to undergo apoptosis. However, Amor-Gueret et al (2006) suggest that BS cells with BLM mutations are able to evade this mechanism by entering into alternative recombination pathways that allow them to progress through the cell cycle even in a hyper-mutated state.
It has been suggested that analysis of BLM protein by immunoblotting and immunohistochemical analysis may be useful in screening for BS, however mutational analysis is required for a concrete diagnosis (Morimoto, 2002). Mutational analysis and genetic testing are important for at-risk couples who are planning to start a family. BS can be diagnosed by the identification of biallelic mutational variants of the BLM gene. However, should this test prove inconclusive; quantifying the frequency of SCE can be used as a further diagnostic tool. BS is the only condition that exhibits this ‘hyper-recombinability’ phenotype, and so such a test can provide a definitive answer (Sanz, 2006).
Because BS is so rare there, is as yet no effective treatment. People with BS should limit their exposure to UV light and should have any infections treated aggressively with antibiotics. Physicians must be mindful of the increased cancer risk and be scrupulous when any potential signs of cancer arise. Frequent screening has also been advised, particularly for colorectal cancer (Arora, 2014). An important consideration when people with BS do develop cancer is how to treat it. Radiotherapy and chemotherapy can increase the likelihood of secondary tumors such as lymphomas, and should be avoided where possible.
BS is an extremely rare condition affecting fewer than 300 people worldwide, with a notable prevalence among the Ashkenazi Jewish community. As there is no effective treatment for the condition, patients and physicians alike must be vigilant and take preventative steps to minimise the risk of developing malignancy. Genetic counselling may also be of benefit to affected individuals for educational purposes and to inform their decision-making if they are considering having children (Cunniff, 2017).
In addition to BLM mutational analysis, knowledge of BLM posttranslational modifications and how they might affect protein function is likely to be an important area of research going forward (Tikoo, 2010). Furthermore, as German et al (2007) identified patients with BS but no BLM mutations, the possibility of heterogeneity underlying BS must be further explored through the use of genome sequencing. Future treatments ought to focus on restoring the proper functioning of BLM, or on inhibition of excessive recombination.
Amor-Gueret, M., (2006) Bloom syndrome, genomic instability and cancer: the SOS-like hypothesis. Cancer Letters, 236(1), pp.1-12.
Arora, H., Chacon, AH., Choudhary, S., McLeod, MP., et al., (2014) Bloom syndrome. International Journal of Dermatology, 53(7), pp.798-802.
Cunniff, C., Bassetti, JA., Ellis, NA,. (2017) Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Molecular Syndromology, 8(1), pp.4-23.
Duker, NJ., (2002) Chromosome breakage syndromes and cancer. American Journal of Medical Genetics, 115(3), 125-9.
Ellis, NA., Groden, J., Ye, TZ., Straughen, J., et al., (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83(4), pp.655-666.
German, J., Roe, AM., Leppert, MF., Ellis, NA., (1994) Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proceedings of the National Academy of Sciences, 91(14), pp.6669-6673.
German, J., Sanz, MM., Ciocci, S., Ye, TZ., et al., (2007) Syndrome-Causing Mutations of the BLM Gene in Persons in the Bloom’s Syndrome Registry. Human Mutation, 28(8), pp.743-753.
Langland, G., Elliott, J., Li, Y., Creaney, J., et al., (2002) The BLM Helicase Is Necessary for Normal DNA Double-Strand Break Repair. Cancer Research, 62(10), pp.2766-2770.
Larizza, L., Roversi, G., Volpi, L., (2010) Rothmund-Thomson syndrome. Orphanet Journal of Rare Diseases, doi: 10.1186/1750-1172-5-2.
Morimoto, W., Kaneko, H., Isoqai, K., Kasahara, K., et al., (2002) Expression of BLM (the causative gene for Bloom syndrome) and screening of Bloom syndrome. International Journal of Molecular Medicine, 10(1), pp. 95-9.
Sanz, MM., German, J., Cunniff, C., (2006) Bloom’s syndrome, n.p.
Tikoo, S., Senqupta, S., (2010) Time to bloom. Genome Integrity, doi: 10.1186/2041-9414-1-14.
Delivering a high-quality product at a reasonable price is not enough anymore.
That’s why we have developed 5 beneficial guarantees that will make your experience with our service enjoyable, easy, and safe.
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more